Asia
Europe, Middle East, Africa
North America










For Clinical Pathogen Outbreak Monitoring
Healthcare Associated Infections (HAI) are common and pose a serious threat to patient safety*.The investigation of possible HAI outbreaks can help track the source of bacterial infections and be used to assist in developing strategies to prevent further spread in the healthcare environment.
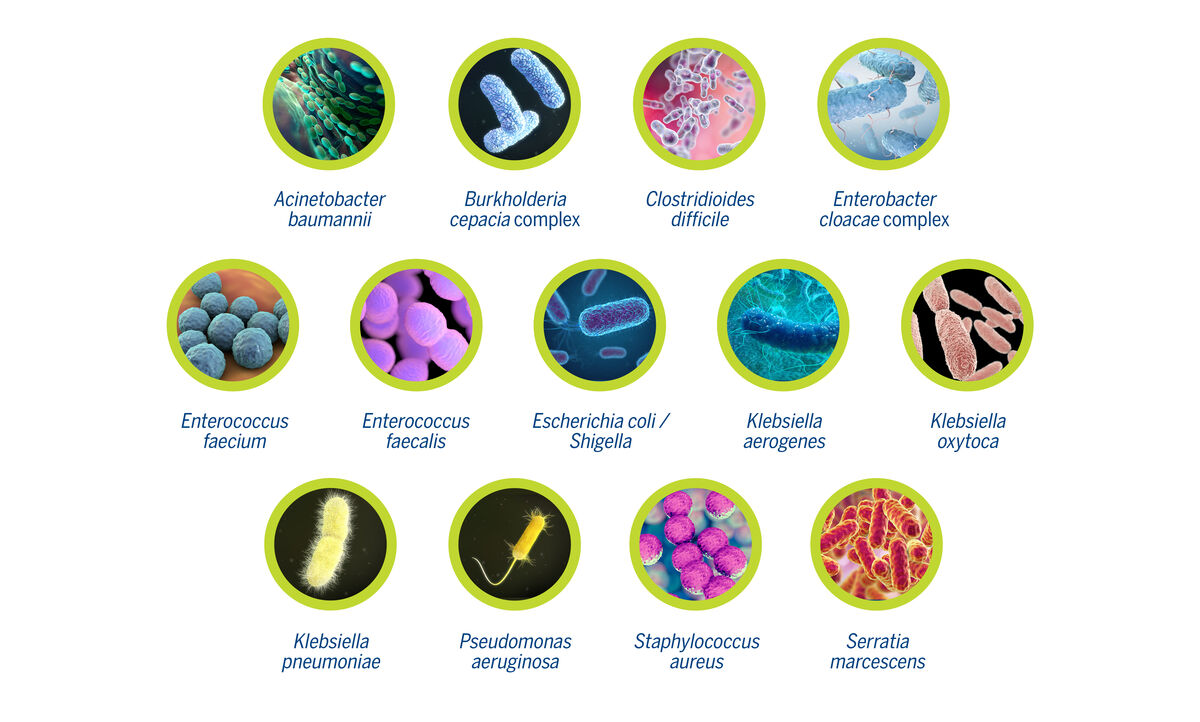
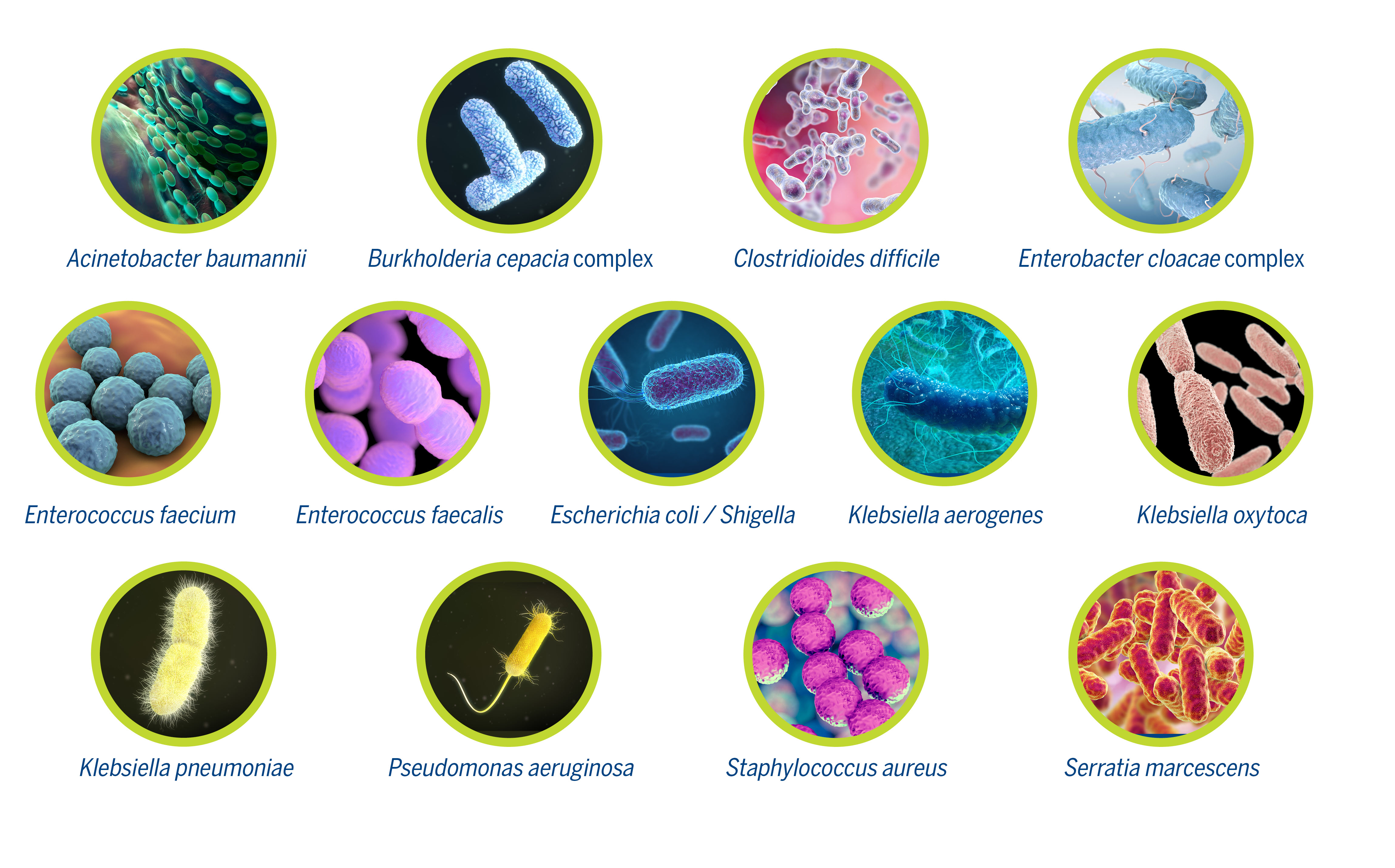
BIOMÉRIEUX EPISEQ CS is a new software as a service application that can help in the management of HAIs in your institution by enabling reliable outbreak investigation decisions. The software tool imports and processes bacterial whole genome sequence data (WGS) for detailed genetic characterization. Accurate determination of bacterial strain relatedness is based on whole genome multi locus sequence typing (wgMLST), providing the highest level of typing resolution. In addition, the presence of resistance and virulence markers are reported, further characterizing the pathogens under investigation. EPISEQ CS for bacterial typing and epidemiology is available for 13 pathogens commonly involved in HAIs.
BIOMÉRIEUX EPISEQ CS puts the power of Next Generation Sequencing into your hands. This software tool automatically imports and processes sequence data for the most common HAI-related organisms, providing accurate determinations of bacterial strain relatedness. In addition, EPISEQ CS reports the presence of resistance and virulence markers.
When Infection Prevention and Control (IPC) recognizes a possible HAI outbreak, EPISEQ CS enables them to partner with the lab to provide actionable answers fast, which helps to confirm the outbreak, track its source, contain the spread, and prevent further infection.
Using Next Generation Sequencing (NGS) and Whole Genome Sequencing (WGS), data from EPISEQ CS can help inform epidemiological decisions by:
In order to protect patients and healthcare workers and to ease the economic burden for hospitals, it’s crucial to quickly recognize HAI outbreaks and pinpoint their source.
Next Generation Sequencing (NGS) and Whole Genome Sequencing (WGS) are the latest molecular technologies with a promising application in high resolution bacterial strain typing. However, routine use of NGS/WGS in clinical microbiology laboratories is hampered by the complexity of bioinformatics tools for the analysis of large amounts of sequence data. EPISEQ CS, a new software as a service application, overcomes these limitations. With one click, EPISEQ CS automatically imports and processes sequence data files and visualizes the relatedness of selected bacterial strains in phylogenetic trees. The cloud-based application puts NGS/WGS-based bacterial typing, along with an extensive knowledge base of more than 50,000 reference strains, at your fingertips. Critical genomic information including resistome and virulome markers are reported to help track, contain, and ultimately stop the spread of infection and prevent HAI.
1. Spinler JK, Raza S, Thapa S, Venkatachalam A, Scott T, Runge JK, Dunn J, Versalovic J, Luna RA, Comparison of Whole Genome Sequencing and Repetitive Element PCR for Multidrug- Resistant Pseudomonas aeruginosa Strain Typing. The Journal of Molecular Diagnostics (2021), doi: https://doi.org/10.1016/j.jmoldx.2021.10.004
| Upload |
Access the cloud-based application and directly upload FASTQ or assembled FASTA sequence files. |
Analyze |
Perform whole genome characterization (wgMLST) and phylogenetic analysis of selected bacterial strains from current outbreaks or compare to historical data from past outbreaks. |
| Report |
Report decisive data to investigate potential outbreak situations including bacterial resistome and virulome information. |
Illumina is a trademark belonging to Illumina Inc.
Whole genome sequencing (WGS) is the latest technology to play an important role in the support of outbreak investigations and epidemiological studies. Accurate bacterial strain typing with WGS has the potential to enhance infection control programs. BIOMÉRIEUX EPISEQ CS uses whole genome sequencing (WGS) and whole genome MLST to analyze thousands of loci across the bacterial genome and identify subtle differences often overlooked by other genotypic methodologies.
bioMérieux has long been a partner to healthcare professionals in the battle against antimicrobial resistance (AMR) and Healthcare Associated Infections (HAI) in particular, offering innovative culture media, identification and susceptibility (ID/AST) reagents and instruments for fast, reliable microbiology testing.
EPISEQ CS now offers a bacterial strain typing application to help you manage HAI in your facilities and provide actionable results. The online tool puts NGS technology, coupled with extensive databases of bacterial strain sequences, at your disposal including:
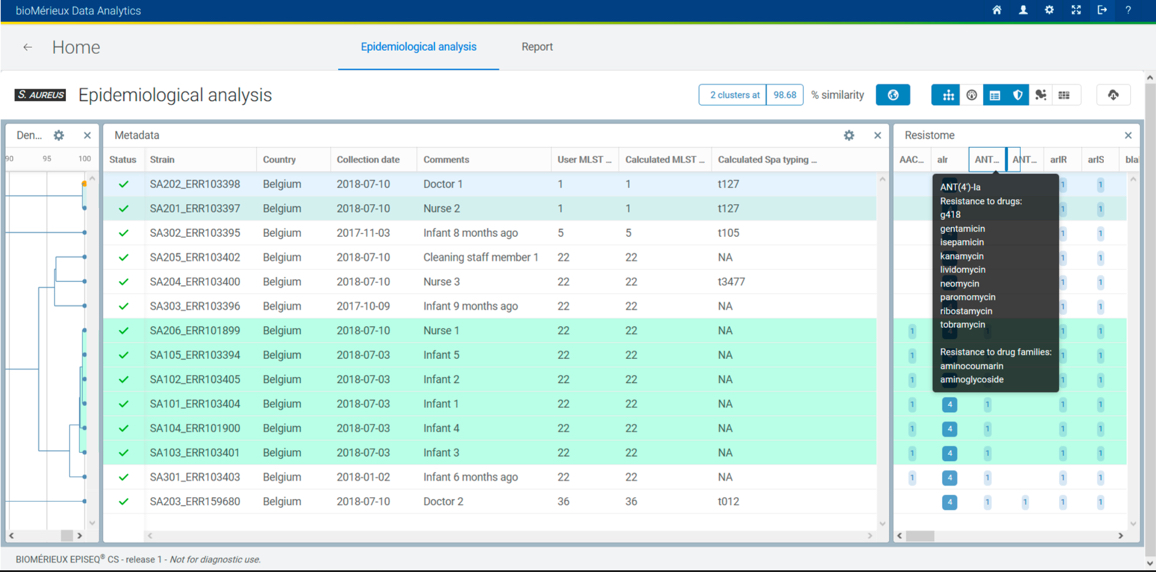
For every sample analyzed in EPISEQ CS, the de novo assembled genome is screened for the presence of wgMLST loci using BLAST. Every locus hit and passing the minimum quality and similarity criteria in the genome is matched against the allele database of the corresponding organism. A called allele has a valid start and stop codon, no internal stop codons, a length divisible by 3, and no non-ACGT bases. Only called alleles are included in the wgMLST allelic profile (i.e., a list of all the called loci) for the sample, which is then used to calculate similarities between the samples. For this, the categorical distance coefficient is used - match or no match between the allele numbers - and UPGMA is used as clustering algorithm. Using this algorithm wgMLST results are then used to:
Easy-to-interpret comprehensive reports in pdf format are automatically created, providing the most precise information at both the individual sample or panel level. These reports document possible strain relatedness and important genetic information of the organisms under investigation.
Phylogenetic analysis based on the wgMLST profiles provides insight into the level of strain relatedness. For each species there are pre-determined similarity thresholds for calling probable or possible relatedness. A historical database for each species is built over time and can be used to compare bacterial strains from new or current outbreaks with those from previous events.
For each individual sample the detailed resistome information is reported including the presence and genomic location of the respective resistant markers (loci) as well as the presence of single (point) mutations.
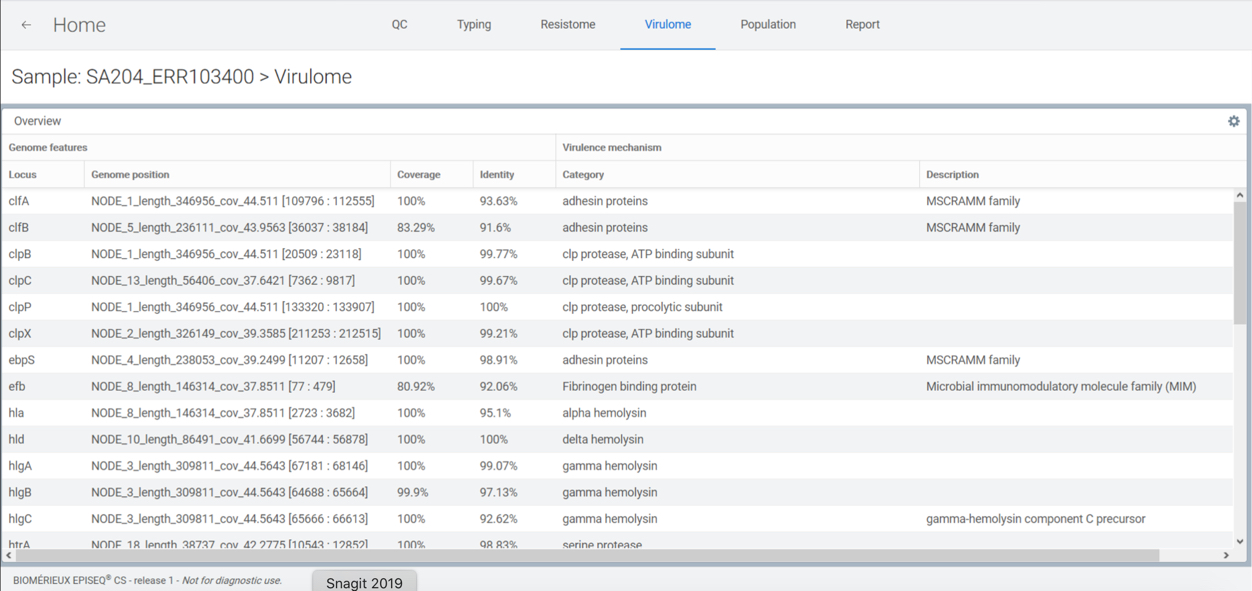
Genomic characterization of the virulome is available for Staphylococcus aureus, Escherichia coli / Shigella spp. and Clostridioides difficile. Any virulence factor detected is reported along with the respective mode of action.
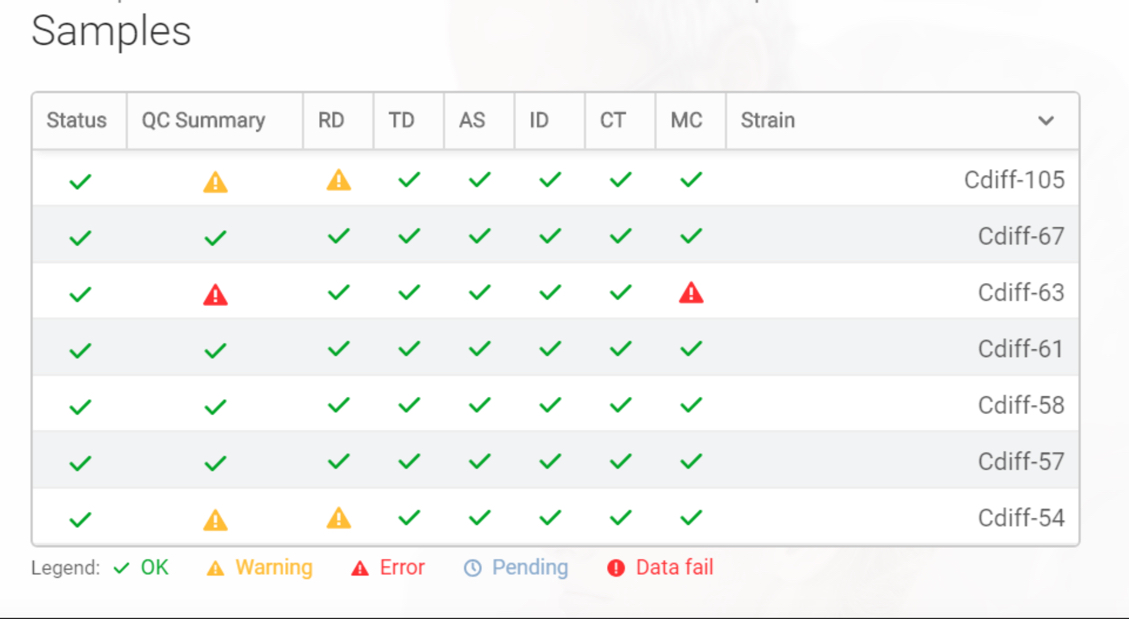
Quality control parameters are calculated for the respective steps in the pipeline for each individual sample. This provides clear visibility and confidence in the quality of the data and the final analysis results. From the general QC summary report, an even more detailed report can be generated.
Each sample is automatically analyzed for strain relatedness against all other samples in the same upload and against the historical database of your previous uploads. The number of probably and possibly related strains is shown in a color coded diagram.
All information obtained in EPISEQ CS can be easily downloaded in pdf format.
PRN 056752 Rev02.A